Background: After viral respiratory infections, like SARS-CoV-2, the risk for cardiovascular complications, including arrhythmias, autonomic dysfunction and heart failure, is significantly increased. Patients with pre-existing cardiometabolic disease are particularly vulnerable and display an elevated risk of developing post-viral cardiovascular complications. To date, insights into how viral infections contribute to the development of cardiovascular pathology remain limited.
Methods: C57BL/6 mice and mice with cardiometabolic disease (LDLR-/- after 8 weeks of high-fat diet) were infected with 1000 plaque-forming units of a mouse-adapted SARS-CoV-2 strain to induce moderate infection severity compared to mock infection. Histology and flow cytometry of cardiac tissue were performed during acute infection (5 days post-infection) and after convalescence (28 days post-infection). Single-nuclei RNA-sequencing (snRNA-seq) of cardiac tissue was performed 28 days after infection.
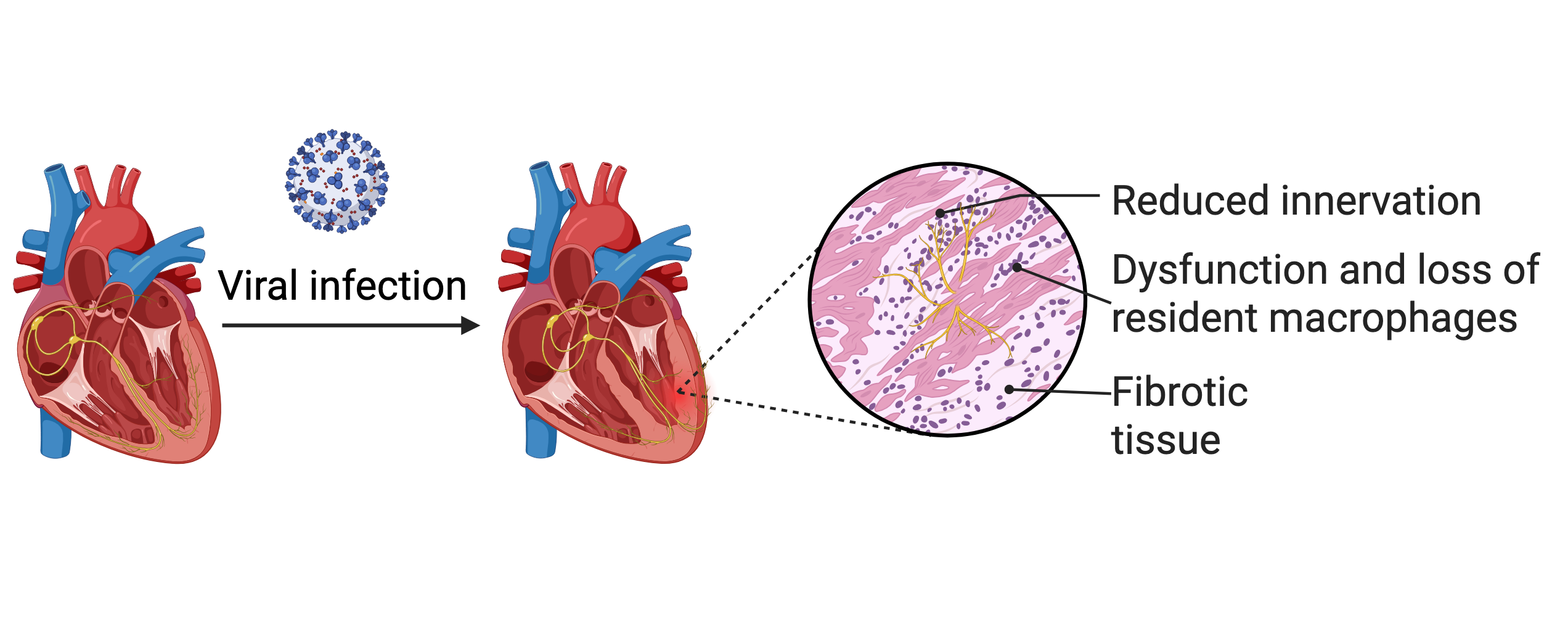
Results: We found that 28 days after SARS-CoV-2-induced viral injury, mice with concomitant cardiometabolic disease (LDLR-/- on a high-fat diet) exhibit amplified cardiac remodelling, including cardiac fibrosis (mock: 0,44±0,03% vs. SARS-CoV-2: 0,65±0.06% sirius red-positive area, p=0.008 and reduced cardiac innervation (mock: 1,0±0,33 vs. SARS-CoV-2: 0,60±0,24 Tuji1+ area, p=0.006). Other histological parameters, such as cardiac hypertrophy and vascularisation, remained unchanged. When examining the immune cell repertoire of the heart after viral injury, we found no increase in recruited myeloid immune cells at either the acute stage or after convalescence. Instead, the number of cardiac tissue-resident macrophages (CCR2-, TIM4+, MHC-II) in cardiometabolic preconditioned animals dropped significantly (mock: 9,2±1,7 vs. SARS-CoV-2: 1,4±0,3 cells/mg heart tissue). Despite partial recovery of this protective macrophage subset, its levels remained markedly reduced at 28 days post-infection. To gain molecular insights into the cellular landscape of the heart following viral injury, we performed snRNA-seq of hearts 28 days after SARS-CoV-2 infection. In resident macrophages, we observed that proinflammatory gene ontology (GO) signatures were upregulated (GO-terms: “antigen processing and presentation of exogenous peptide”, “positive regulation of immune response”). At the single-gene level, we found upregulation of genes associated with MHC-II antigen presentation and pro-inflammatory pathways, including complement factors and cathepsins. Notably, not only were proinflammatory pathways enriched, but genes associated with homeostatic function were downregulated, as reflected by the downregulated GO-terms “blood vessel development”, “heart development” and “axon guidance”. In addition, in cardiac fibroblasts, we describe upregulation of GO-terms such as “supramolecular fibre organisation” in line with the observed diffuse cardiac fibrosis after viral injury.
Conclusions: Our data provides mechanistic insights into cardiovascular complications after viral infection. Since no replicable virus or viral RNA was detected in the heart at either the acute or the recovery stage, our data suggest that secondary inflammatory processes are responsible for the observed phenotype.