Background:
Rare variants in cardiomyopathy (CMP) genes have been linked to adverse cardiovascular outcomes, highlighting the potential of genome-first approaches to identify risk beyond traditional family screening. Most prior studies have been restricted in size or focused on European ancestry populations, limiting understanding of how these variants affect disease expression in unselected adults.
Objective:
To characterize the prevalence and clinical consequences of rare pathogenic CMP gene variants in large, population-based cohorts.
Methods:
Exome and genome sequencing data from the UK Biobank (UKB) and the All of Us Research Program (AllofUs) were used to identify carriers of pathogenic or likely pathogenic variants in genes with strong evidence for dilated, hypertrophic, or arrhythmogenic cardiomyopathy. Analyses included ClinVar-annotated variants and predicted loss-of-function variants with established disease mechanisms. Time-to-event models were applied to estimate associations between carrier status and incident cardiovascular outcomes, including heart failure, non-ischemic cardiomyopathy, atrial fibrillation, and ventricular tachycardia.
Results:
The study included 392,224 UKB participants (mean age 57 years; 54% women) and 193,222 AllofUs participants (mean age 52 years; 61% women). The prevalence of rare CMP variants ranged from 1.06% to 1.40%. Across both cohorts, carriers had a significantly higher risk of cardiovascular events compared with noncarriers. In UKB participants, the hazard ratio (HR) for the composite cardiovascular outcome was 1.75 (95% CI 1.62–1.90; p<2x10-15). In AllofUs, the corresponding HR was 1.95 (1.74–2.18; p<2x10-16). By age 75 years, cumulative incidence of the cardiovascular composite outcome was 24.9% in carriers versus 14.0% in noncarriers in the UKB, and 34.5% versus 18.0% in AllofUs. Associations were consistent for arrhythmic events and across gene-level analyses. Notably, TTN-rare variant carriers across all reportable ancestry subgroups had an increased risk of all individual and composite clinical outcomes.
Conclusion:
In two large population-based cohorts totaling more than half a million participants, rare pathogenic CMP gene variants were present in approximately 1% of adults and were associated with a substantially increased risk of cardiomyopathy, arrhythmia, and heart failure. These findings demonstrate the clinical relevance of rare CMP variants in unselected populations and support the integration of genomic information into cardiovascular risk assessment at scale.
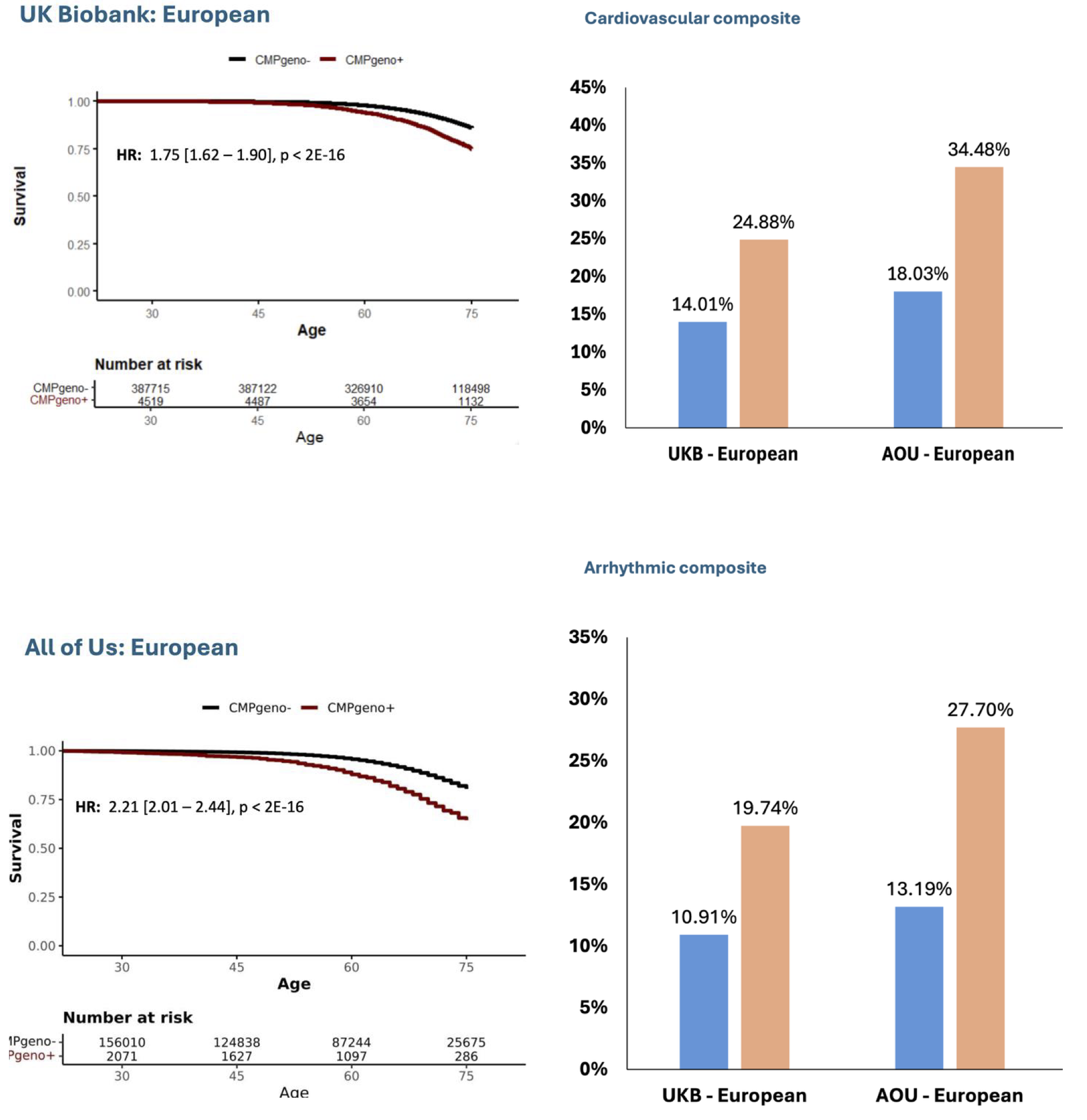
Figure 1: Kaplan-Meier curves of event-free survival in carriers (CMPgeno+, red) and non-carriers (CMPgeno-, blue). On the right, cumulative incidence by age 75 between carriers (CMPgeno+, organe) and non-carriers (CMPgeno-, lightblue).



