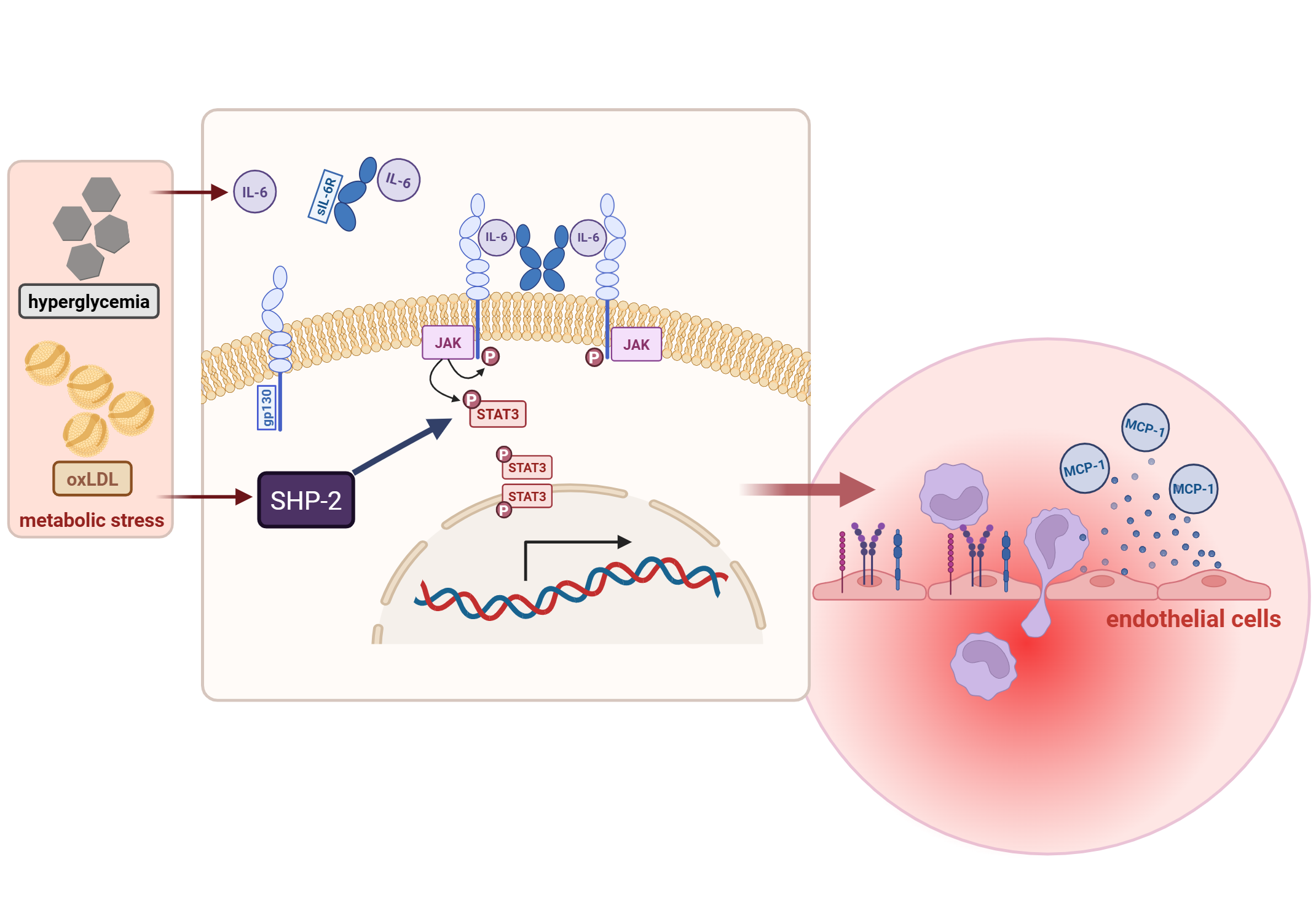
Background/Purpose: Interleukin-6 (IL-6) levels serve as established biomarker for cardiovascular diseases such as coronary artery disease and atherosclerosis. Although IL-6 signalling contributes to vascular inflammation and remodeling, its role in atherosclerosis remains controversial. Emerging evidence suggests that distinct IL-6 signaling pathways exhibit opposing effects: IL-6 trans-signalling (IL-6 TS) via soluble IL-6R (sIL-6R) drives pro-inflammatory vascular responses, whereas classic signalling promotes protective and reparative functions. The tyrosine phosphatase SHP-2 regulates JAK/STAT signalling; however, its role in controlling IL-6 trans-signalling remains unclear. This study investigates how SHP-2 modulates IL-6 trans-signalling-dependent endothelial activation.
Methods: Human umbilical vein endothelial cells (HUVECs) from pooled donors (n = 4) were used to investigate IL-6–dependent signalling and endothelial activation. IL-6 trans-signalling was induced by stimulating HUVECs with IL-6 +sIL-6R, in the presence or absence of pharmacological inhibitors targeting SHP-2 or STAT3. Downstream signalling events were assessed by Western blotting using phospho-specific antibodies for STAT3 (pSTAT3). MCP-1 secretion was quantified by ELISA. Inflammatory gene expression and adhesion molecule levels (ICAM-1, VCAM-1, E-Selectin, JAM-1, PECAM-1) were determined by qPCR and flow cytometry. Functional endothelial responses were assessed using transwell and flow-based monocyte adhesion and transmigration assays. To model metabolic stress associated with cardiometabolic disease, cells were exposed to either oxidized low-density lipoprotein (oxLDL) or a high-glucose/methylglyoxal (MG/HG) microenvironment, and SHP-2 protein expression was quantified by Western blotting.
Results: IL-6 TS markedly increased MCP-1 secretion (p=0.0025); pharmacological inhibition of SHP-2 further amplified MCP-1 production (p=0.0001). Co-treatment with a STAT3 inhibitor significantly reduced MCP-1 secretion, demonstrating STAT3-dependent regulation. Correspondingly, qPCR and flow cytometry analyses revealed that IL-6 TS upregulated adhesion molecules including VCAM, E-Selectin, JAM-1, and PECAM; SHP-2 inhibition further enhanced their expression. Functional relevance was confirmed using monocyte–endothelial interaction assays. Under IL-6 TS conditions, SHP-2 inhibition significantly enhanced monocyte adhesion (p=0.001) and transmigration (p=0.0038) across endothelial monolayers. STAT3 inhibition reversed these effects, significantly decreasing both monocyte adhesion (p=0.001) and transmigration (p=0.0009). Metabolic stress mimicking hyperglycemia and hypercholesterolemia increased endothelial IL-6 release and elevated SHP-2 expression, linking metabolic activation to altered IL-6 signalling.
Conclusions: SHP-2 phosphatase acts as a critical negative regulator of IL-6 trans-signalling in endothelial cells. SHP-2 inhibition enhances STAT3-dependent inflammatory signalling, adhesion molecule expression, and monocyte recruitment. These findings identify SHP-2 as a key checkpoint limiting IL-6-driven endothelial inflammation and establish it as a potential therapeutic target for reducing vascular inflammation in cardiometabolic disease.