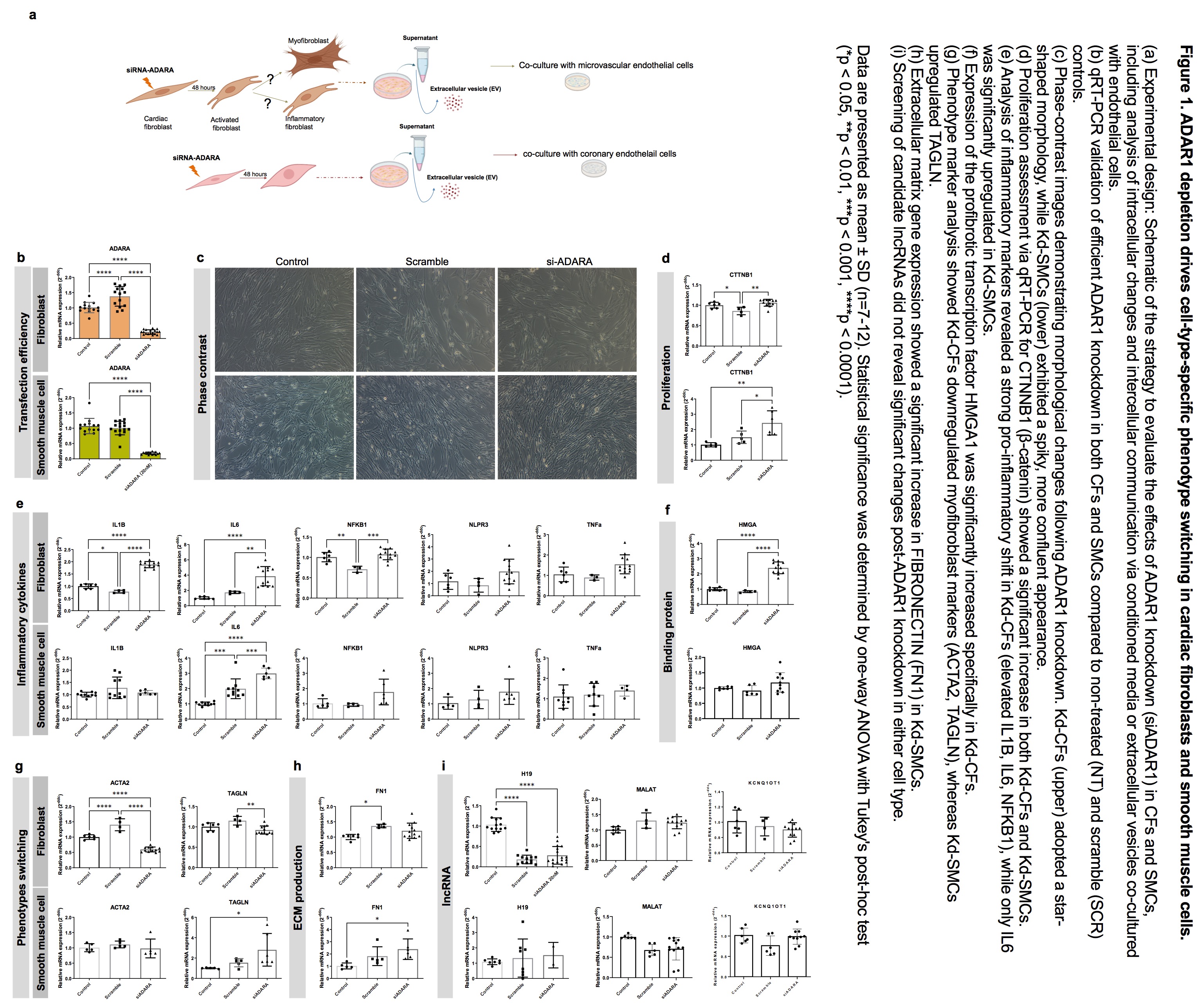
Rationale: Aortic valve stenosis (AVS) and coronary artery disease (CAD) represent a major clinical synergy, yet their shared molecular drivers remain elusive. While pathological phenotype switching of cardiac fibroblasts (CFs) and smooth muscle cells (SMCs) is a hallmark, the upstream regulators are poorly defined. We identify a novel axis: RNA editing by ADAR1. This enzyme installs millions of A-to-I modifications, acting as a master epitranscriptomic regulator. The direct genetic link between human ADAR1 mutations and AVS provides a compelling, yet unexplored, pathogenic mechanism. We hypothesize that ADAR1 serves as a critical brake on phenotype switching, and its loss unleashes cell-specific programs that drive both AVS and CAD from a shared molecular origin.
Methods and Results: Using siRNA-mediated knockdown in primary human CFs and SMCs, we achieved efficient ADAR1 depletion, confirmed by qRT-PCR. We discovered that ADAR1 loss triggers strikingly distinct, lineage-locked phenotypic transitions. CFs underwent a profound morphological and functional shift, abandoning their classic myofibroblast destiny (evidenced by downregulation of ACTA2 and TAGLN) to adopt a novel, highly inflammatory state. This was marked by a dramatic surge in IL1B, IL6, and NFKB1, coupled with upregulation of a pro-fibrotic signaling hub (HMGA1, HIF1A, ELAVL1). In stark contrast, SMCs responded to the same genetic insult by hyper-proliferating (increased CTNNB1) and adopting a synthetic phenotype, characterized by enhanced ECM production (elevated FN1 and TAGLN) without a significant inflammatory response. This cell-type-specific decoding of the same epitranscriptomic signal is a novel and fundamental finding.
Conclusion: We pioneer the discovery that ADAR1 is a master regulator of cardiovascular cell fate, whose loss initiates divergent pathogenic pathways from a common origin. We reveal a novel inflammatory fibroblast phenotype in AVS and a distinct synthetic SMC program in CAD, both orchestrated by loss of a single RNA-editing enzyme. This work fundamentally shifts the paradigm by positioning the epitranscriptome and A-to-I editing as a central, previously unrecognized layer of regulation in common cardiovascular diseases, unveiling the ADAR1 pathway as a entirely new therapeutic frontier for precise, mechanism-based intervention.