Background:
Inflammation is a major cardiovascular risk factor that promotes atherothrombosis. Pharmacological inhibition of the IL-1β-IL6 axis has proven effective in reducing atherothrombotic events, suggesting that anti-inflammatory therapy may exert antithrombotic effects. Platelets play a central role in this process. However, the extent to which anti-inflammatory interventions modulate platelet reactivity remains uncertain.
Aim:
To assess the relationship between inflammation and platelet reactivity in participants of the Ludwigshafen Risk and Cardiovascular Health (LURIC) study.
Methods and Results:
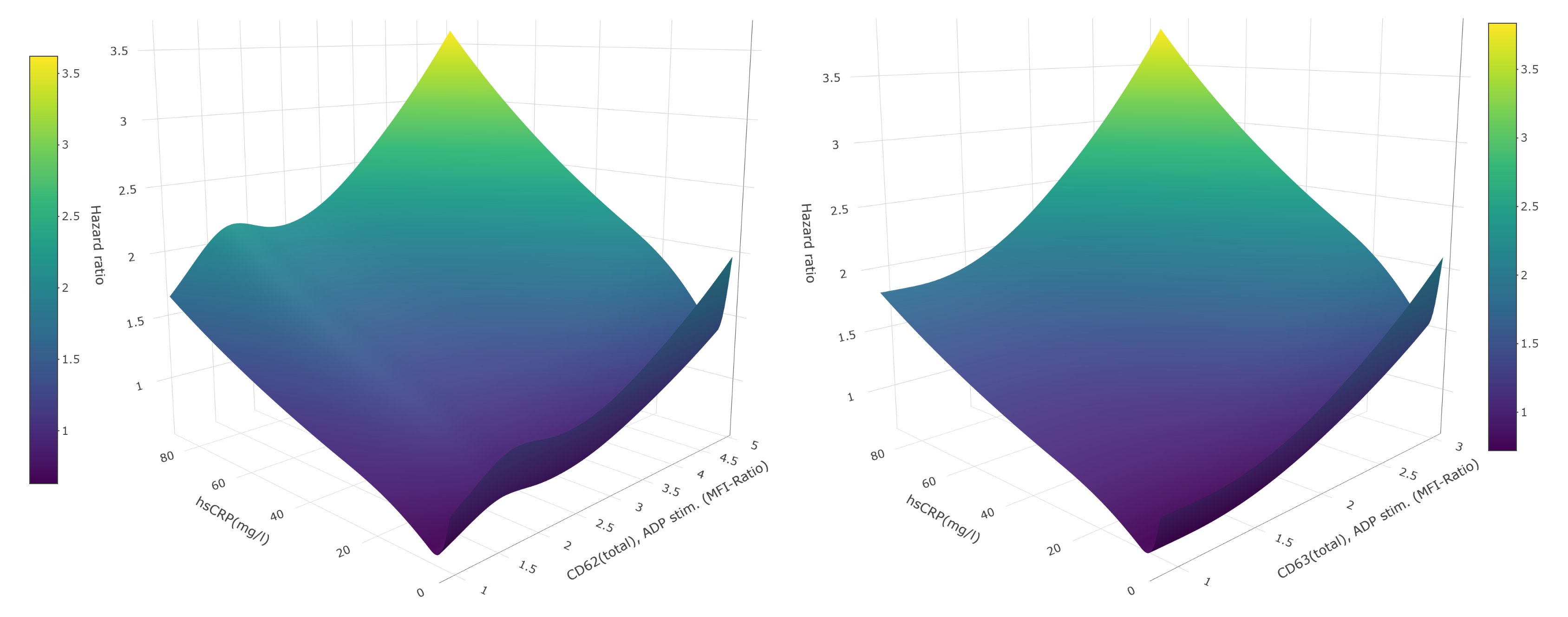
Platelet reactivity and hsCRP data (CD62p, CD63, and fibrinogen binding after ADP-stimulation) were available from 1,242 patients undergoing coronary angiography between 1997 and 2000. Patients within the highest hsCRP tertile exhibited a markedly adverse cardiovascular risk profile, including higher prevalences of myocardial infarction, stroke, atrial fibrillation, and type 2 diabetes mellitus (all p < 0.05). Higher inflammation was associated with significantly increased ADP-induced platelet activation, as reflected by elevated CD62p and CD63 expression in the highest vs. lowest hsCRP tertile (CD62p: 1.76 [95% CI 1.49–2.20] vs. 1.89 [95% CI 1.56–2.46], p < 0.01; CD63: 1.27 [95% CI 1.15–1.43] vs. 1.34 [95% CI 1.17–1.59], p < 0.01). No associations were observed for ADP-induced fibrinogen binding. Both ADP-induced CD62p and CD63 expression independently predicted mortality in age- and sex-adjusted cox regression models (CD62p: HR 1.20 [95% CI 1.02–1.42], p = 0.028; CD63: HR 1.21 [95% CI 1.08–1.35], p = 0.001). In addition, continuous restricted cubic spline analyses confirmed a strong, continuous association between platelet reactivity, inflammation (i.e. hsCRP), and mortality (Figure 1). To explore whether IL-6–dependent inflammation causally influences platelet reactivity, we examined the IL-6–signalling modulating SNP rs2228145 (A>C), known to reduce IL-6 pathway activity and hsCRP levels. Homozygosity for the C-allele significantly attenuated mortality risk in patients with heightened ADP-induced platelet activation (CD62p: HR 0.55 [95% CI 0.35–0.88], p = 0.013; CD63: HR 0.61 [95% CI 0.39–0.96], p = 0.033). However, homozygous presence of the C-allele variant did not affect ADP-induced platelet reactivity itself. These findings indicate that although impaired IL-6 signalling mitigates mortality in individuals with heightened platelet activation, IL-6-mediated inflammation does not directly modulate platelet reactivity.
Discussion:
Our data demonstrate a robust association between inflammation, ADP-induced platelet activation, and mortality within the LURIC cohort. Nonetheless, genetic impairment of IL-6 signalling does not influence platelet reactivity, suggesting that IL-6–driven inflammation does not causally regulate ADP-induced platelet activation.
Figure 1: Association between ADP-induced CD62p and CD63 platelet reactivity, inflammation (i.e. hsCRP) and mortality