Background:
Mitochondrial dysfunction contributes to impaired myocardial energetics and diastolic performance in heart failure with preserved ejection fraction (HFpEF). The mitochondria-targeted peptide Elamipretide has shown encouraging effects on myocardial energetics and function in preclinical heart failure models and early clinical studies in HFrEF, yet data in HFpEF remain limited. Whether targeting mitochondrial dysfunction can confer measurable cardiac benefit in HFpEF remains to be determined.
Methods:
Female obese ZSF1 rats with established HFpEF received either NaCl (HFpEF) or Elamipretide for 12 weeks (HFpEF/Ela), with age-matched female lean ZSF1 rats serving as healthy controls (con). Cardiac function and hemodynamics were assessed by echocardiography and invasive measurements. Left-ventricular (LV) mitochondrial function was evaluated in saponin-permeabilized fibers, and mitochondrial ultrastructure was analyzed by Transmission Electron microscopy. Molecular and histological analyses included cardiolipin profiling and expression analyses of markers of hypertrophy, fibrosis, and inflammation. Carotid vascular reactivity and stiffness were measured ex vivo.
Results:
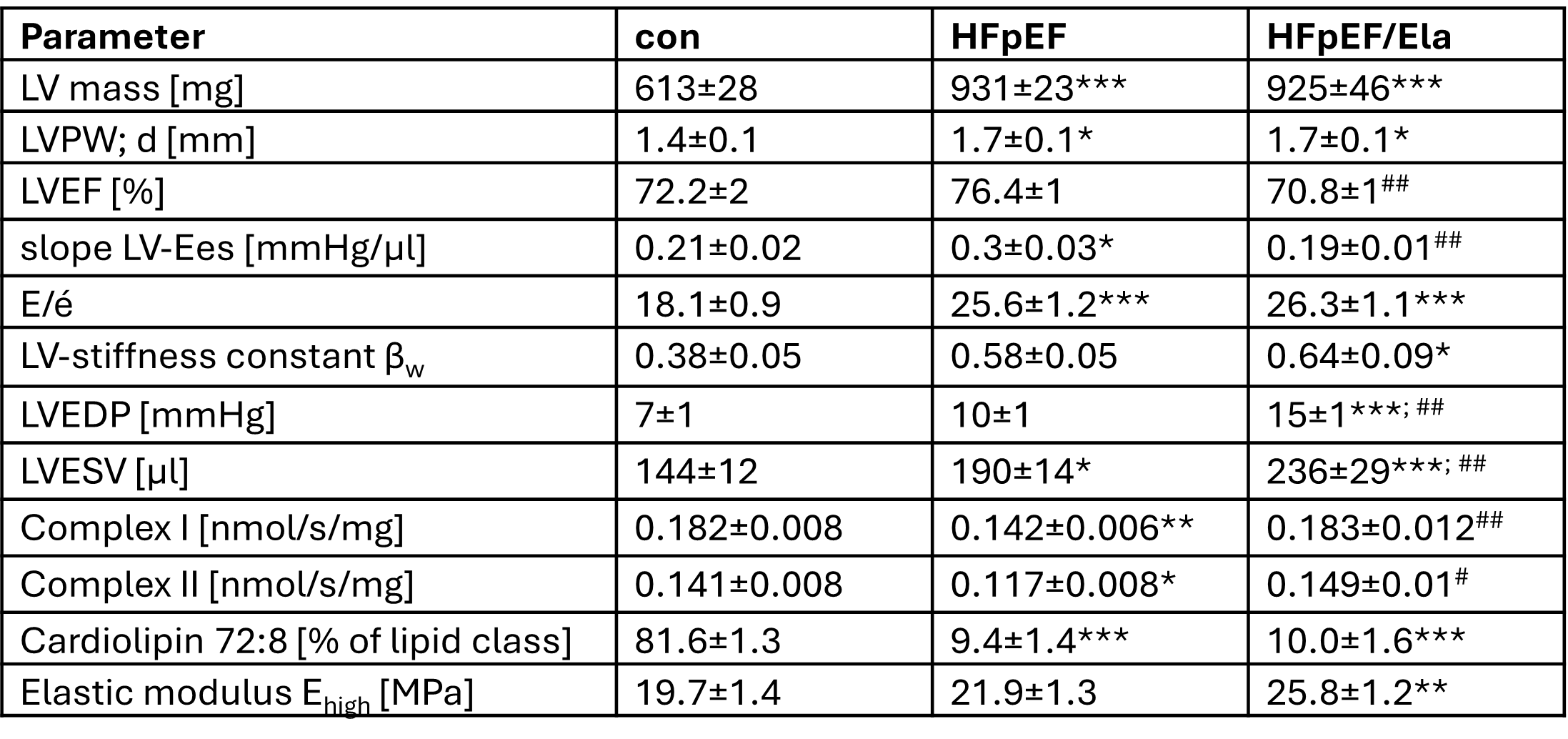
Elamipretide modestly enhanced mitochondrial respiration via complexes I and II in LV fibers, while mitochondrial ultrastructure, cardiolipin (72:8) content, and tafazzin expression remained unchanged. Diastolic dysfunction in HFpEF persisted under treatment, as reflected by elevated E/e′. Ventricular stiffness, assessed by stiffness constant β and titin phosphorylation, remained comparable to untreated HFpEF. Systolic performance was slightly impaired, with modest reductions in LV ejection fraction and slope LV Ees, indicating a mild decrease in contractility. Hypertrophic remodeling, reflected by reduced Myh6/Myh7 ratio and increased wall thickness, was evident in HFpEF and unaffected in HFpEF/Ela. Profibrotic gene expression was markedly elevated in HFpEF and remained unchanged with treatment. Myocardial inflammation appeared modestly enhanced in HFpEF/Ela compared to HFpEF as reflected by increased Cd68 expression. Although carotid stiffness (elastic modulus Ehigh) increased under Elamipretide, arterial function and morphology were unaltered.
Conclusions:
In experimental HFpEF, mitochondrial targeting by Elamipretide enhanced mitochondrial respiration but failed to restore cardiac function or reverse remodeling, suggesting that modulation of mitochondrial bioenergetics alone may be insufficient once HFpEF is established. These findings underscore the need for further studies to evaluate earlier intervention, combination therapies, or alternative strategies to determine whether mitochondrial targeting can provide functional benefit in HFpEF.
Table 1. Animal characteristics. *p<0.05, **p<0.01, ***p<0.001 vs. con, #p<0.05, ##p<0.01 vs. HFpEF