Introduction:
Heart failure with preserved ejection fraction (HFpEF) accounts for nearly half of all heart failure cases and remains a leading cause of morbidity and mortality worldwide. Growing evidence suggests that HFpEF represents a multisystemic metabolic disorder rather than an isolated cardiac disease, which likely contributes to the remaining lack of causally directed pharmacotherapies. Recent large cohort studies have demonstrated that metabolic dysfunction-associated fatty liver disease (MAFLD) is independently associated with increased incidence and severity of HFpEF. Conversely, impaired cardiac function may aggravate hepatic congestion and fibrosis, emphasizing a bidirectional relationship. Understanding the liver–heart interorgan crosstalk may reveal mechanistic drivers of HFpEF and uncover novel therapeutic targets.
Methods:
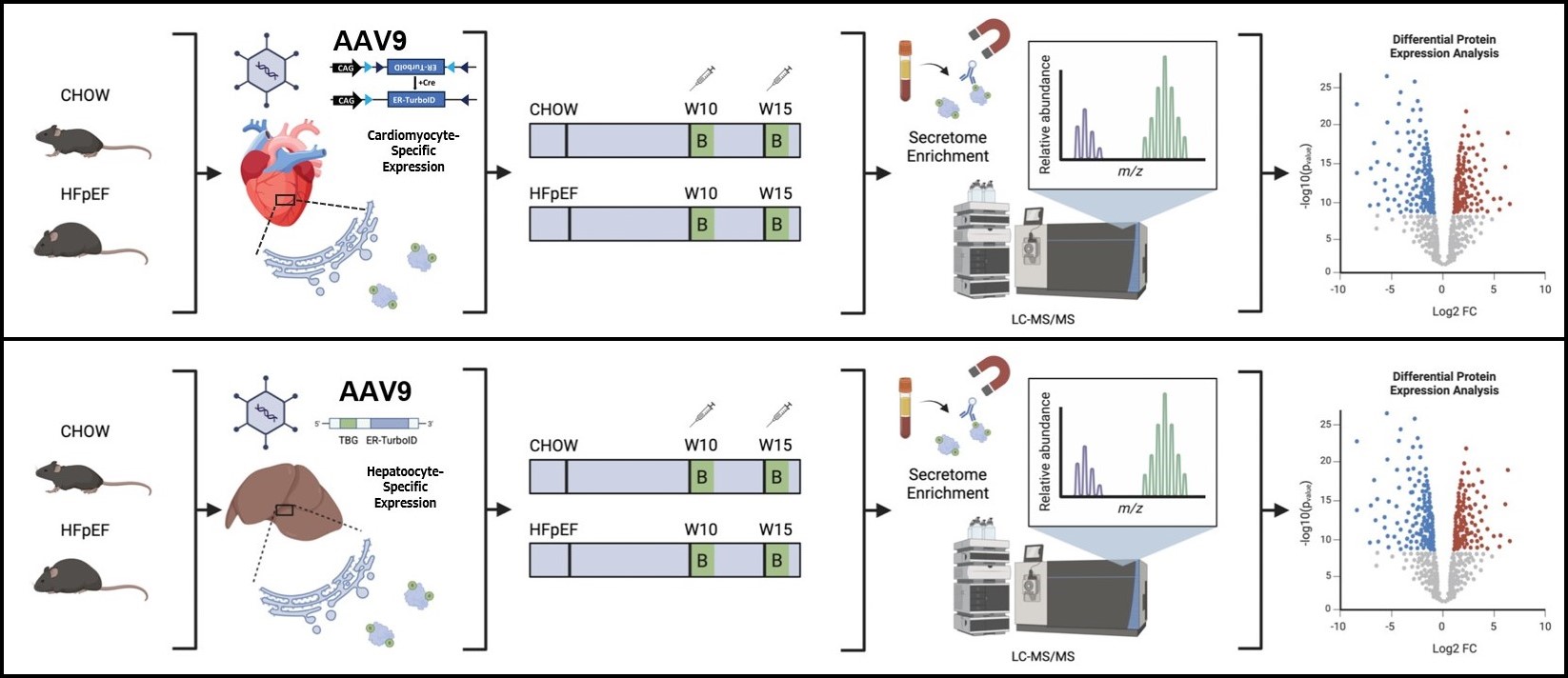
To explore the interorgan crosstalk in cardiometabolic HFpEF, we used an innovative in vivo proximity labeling strategy enabling cell type–specific secretome mapping. Proteins trafficking through the endoplasmic reticulum (ER) on the classic secretory pathway were covalently biotin-tagged by the ER-resident biotin ligase TurboID. Hepatocyte-specific TurboID expression was achieved using an AAV9 vector carrying a thyroxine-binding globulin (Tbg) promoter, while cardiomyocyte-specific expression was obtained via Cre-dependent inversion in αMHC-Cre mice. HFpEF was induced using a “two-hit” model combining a high-fat diet with nitric oxide synthase inhibition by L-NAME. Biotin was administered intraperitoneally for three consecutive days at defined time points during disease progression. Biotinylated proteins were isolated from plasma or tissue lysates using streptavidin magnetic beads and analyzed by LC-MS/MS. Phenotypic characterization included echocardiography, metabolic testing, histology, and gene expression profiling by qRT-PCR.
Results:
Successful hepatocyte- and cardiomyocyte-specific expression and enzymatic activity of TurboID were confirmed by immunostaining and Western blotting. Quantitative proteomics revealed distinct organ-specific secretomes under HFpEF conditions, including differential regulation of multiple hepatocyte-derived plasma proteins compared with chow-fed controls. Notably, proteins labeled in the liver were detectable not only in plasma but also in heart, muscle, and adipose tissue lysates, indicating active interorgan transfer of secreted factors. These findings validate the feasibility and precision of in vivo organ-specific secretome profiling to delineate systemic signaling pathways in cardiometabolic HFpEF.
Conclusion:
This study establishes a technically robust in vivo approach for organ- and cell type–specific secretome analysis in a murine HFpEF model. The ability to trace secreted proteins from their tissue of origin to target organs provides novel insights into the molecular crosstalk underlying this multisystemic disease. This spatially resolved proteomic framework offers mechanistic insight into the pathophysiology of HFpEF and establishes a foundation for the identification of circulating effectors and therapeutic targets.



