Introduction: Congenital Long-QT-Syndrome (LQTS) is a hereditary channelopathy characterized by delayed cardiac repolarization, associated with an increased risk of syncope and sudden cardiac death (SCD). Its diagnosis can be challenging in patients with coexisting heart diseases that may mask clinical symptoms or electrocardiographic parameters. We report a case of a woman with a congenital atrioventricular septal defect (AVSD), Wolff-Parkinson-White (WPW) syndrome, and recurrent syncope, in whom LQTS was only recognized several years after initial evidence of QT prolongation.
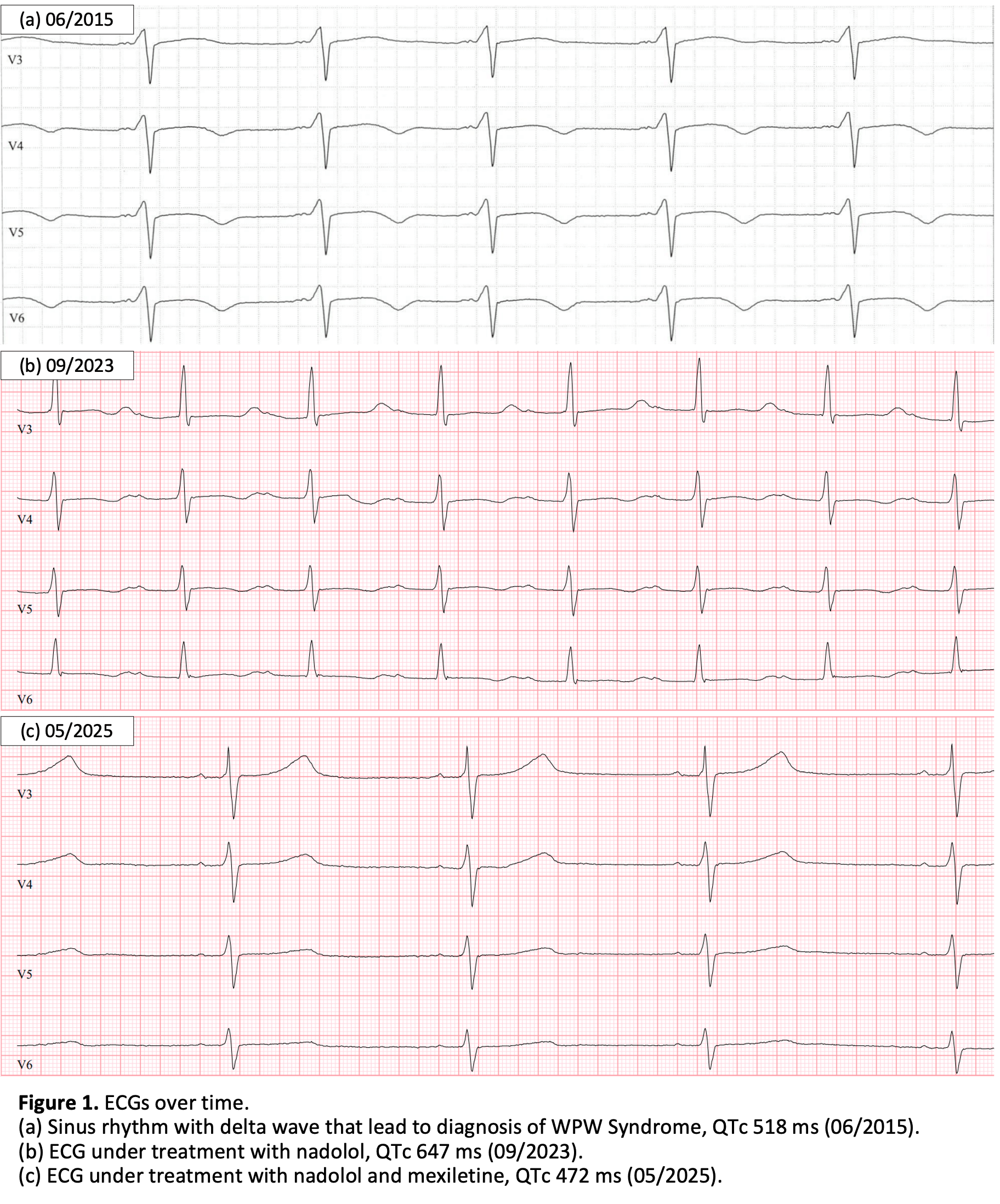
Case Description: The patient (female) was born in 2001 with an AVSD and underwent surgical correction in early childhood. She recovered and demonstrated normal growth with no abnormalities in the following echocardiographic exams. At the age of 14 years, she was diagnosed with WPW Syndrome, after a routine ECG demonstrated delta waves in leads aVL and V2-V6. Later, she reported recurrent episodes of dizziness, for which beta-blocker therapy with metoprolol was started. Treatment was discontinued after symptom resolution, but the patient underwent successful catheter ablation of an anterior accessory conduction pathway. At the age of 22 years, the patient presented to the emergency room after suffering from recurrent syncope. The surface ECG demonstrated a wide-complex tachycardia with delta waves, suggesting recovery of the accessory pathway leading to repeated ablation. Post-ablation, delta waves were no longer present, but a QTc interval of 510 ms was observed. This marked the first active recognition of QTc prolongation in this patient, leading to the initiation of genetic testing, which revealed a heterozygous pathogenic variant in the KCNH2 gene (NM_000238.3:c.2987A>T p.(Asn996Ile)). Even under pharmacological treatment with propranolol and later nadolol, QTc intervals remained prolonged with a maximum of 690 ms. After additional therapy with mexiletine was initiated, a shortening of the QTc interval to <500 ms was observed.
Retrospectively, the first abnormal surface ECG dates back to 2003, with borderline prolonged QTc interval of 460 ms, followed by the first definite documentation of a prolonged QTc of 520 ms in 2015. Although the patient was followed up regularly, prolonged QTc intervals were not recognized and no suspicion of LQTS was raised until recently.
Discussion: This case illustrates how significant QTc prolongation in multiple ECGs can be overlooked for years in patients when symptoms are attributed to other cardiac conditions. The coexistence of AVSD and WPW syndrome may have masked or confounded early diagnosis of LQTS. This highlights the importance of a thorough ECG review with manual QTc measurement in patients with recurrent syncope and may prevent serious arrhythmia episodes leading to syncope or sudden cardiac death in patients and their families.