Background: Connexin 43 (Cx43), a gap junction forming hexameric transmembrane protein located at the intercalated discs of cardiomyocytes (CM), plays a key role in coordinated electrical excitation and subsequent mechanical contraction. Beyond its well-known canonical role, Cx43 is also located in CM mitochondria, which are critical for CM function and survival during myocardial ischemia/reperfusion (I/R). Since increased expression and/or translocation of endogenous Cx43 to mitochondria and its phosphorylation reduce I/R injury, overexpression of Cx43 in CM could potentially provide cardioprotection through such non-canonical action of Cx43. However, constitutive Cx43 overexpression models caused developmental defects and reduced postnatal viability in mice, and the adeno-associated virus-mediated Cx43 overexpression models lack cell- and tissue-specificity, which complicates the interpretation of results.
Aim: Generation of tamoxifen (Tx)-inducible CM-specific Cx43 overexpression (iCx43OE-αMHC) mice to characterize the cardioprotective potential of CM-specific Cx43 overexpression with the focus on infarct size development and mitochondrial function.
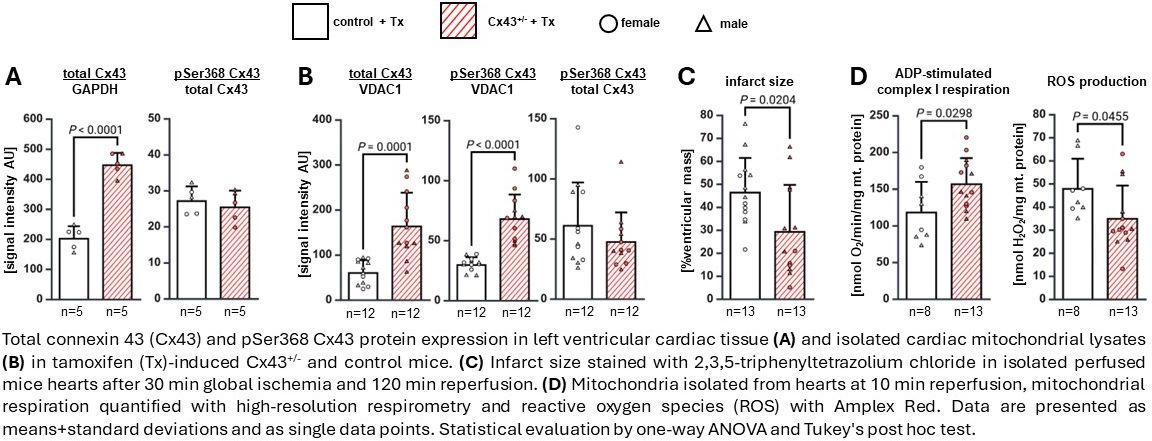
Methods and Results: Our transgenic mouse line with conditional Cx43 overexpression in cardiomyocytes (iCx43OE-αMHC) expressed murine Cx43 with a C-terminal P2A residue and the reporter mCherry in CM upon Tx-induction. All analyses were performed 14 days after Tx induction; iCx43OE-αMHC mice were viable and displayed no overt phenotype. The overexpression of Cx43 and its prototypic phosphorylation (pSer368) was analyzed in left ventricular (LV) tissue from Cx43+/- and littermate control mice by Western blotting. Total Cx43 protein expression was increased in Cx43+/- hearts, and the transgenic Cx43 was proportionally phosphorylated to the intrinsic Cx43 (Fig. A). Mitochondria were isolated by differential centrifugation from naïve hearts of Cx43+/- or control mice. Total and phosphorylated Cx43 protein levels were enhanced in isolated mitochondria of Cx43+/- hearts, indicating increased translocation of Cx43 to the mitochondria in transgenic hearts (Fig. B). Mitochondrial respiration was measured using substrate-inhibitor-uncoupler-titration high-resolution respirometry. Baseline, ADP-stimulated complex I, uncoupled, complex II, and complex IV respiration in mitochondria from Cx43+/- hearts did not differ from those in controls (data not shown). To assess whether the transgenic Cx43 overexpression provides cardioprotection, isolated perfused hearts from control and Cx43+/- mice were subjected to 30 min of global ischemia followed by 120 min of reperfusion and coronary flow and LV-developed pressure measured. Cx43+/- hearts were protected, as coronary flow and LV developed pressure were better restored during reperfusion, and consistent with this finding, infarct size was reduced by 37% (Fig. C). In mitochondria isolated after 10 min of reperfusion from Cx43+/- mice, mitochondrial function was improved, as ADP-stimulated complex I respiration was increased and ROS production decreased (Fig. D).
Conclusion: While transgenic Cx43 overexpression does not impact mitochondrial function under naïve conditions, it enhances mitochondrial function during cardiac ischemic stress, providing a potentially novel cardioprotective strategy.