IntroductionCardiometabolic diseases such as heart failure with preserved ejection fraction (HFpEF) and atherosclerosis are among the leading causes of mortality worldwide. Whereas the enzymatic activation of histone deacetylase 4 (HDAC4) plays a significant in the pathogenesis of HFpEF, there is strong genetic and experimental evidence that HDAC9 is critical for the development of atherosclerosis. The role of the other class IIa HDACs (4, 5 and 7) for atherosclerosis has not been investigated but is important with respect to the development of HDAC inhibitors. This study tries to shed light on the role of HDAC4 in an atherosclerotic mouse model.
MethodsMale adult mice with an enzymatically deficient mutant of HDAC4 (HDAC4ki) and wildtype controls (Crtl) were subjected to a non-genetic metabolic atherosclerosis (ASK) protocol consisting of an AAV8 injection delivering a gain-of-function mutation of proprotein convertase subtilisin/kexin type 9 (PCSK9) resulting in the induction of hypercholesterinaemia, a 5-day treatment of Streptozotocin (STZ) to induce diabetes mellitus type 1 and a 12-week Western diet. In the last week of the study, systolic and diastolic heart function was assessed via echocardiography and blood pressure was measured non-invasively on the tail. During organ harvest, the aortic root and arch were dissected to analyse the extent and quality of atherosclerotic plaques.
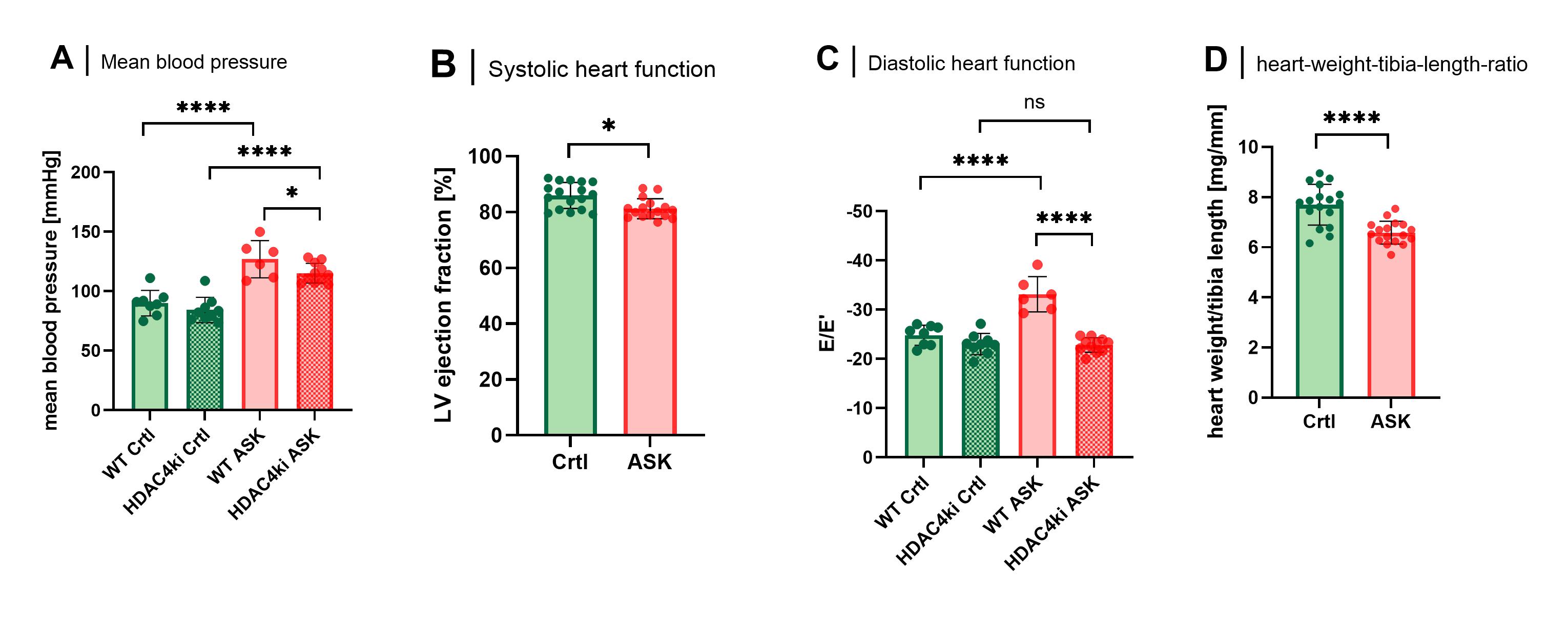
ResultsHypercholesterinaemia and diabetes were successfully implemented noticeable in a more than 15-fold increase of total cholesterol levels 4 weeks after AAV injection and a mean blood glucose level of 512 mg/dl in the ASK mice. A higher mean blood pressure could be measured in the ASK group (ASK all: 119.3 ± 12.37 mmHg, n=17; Crtl all: 86.99 ± 10.78 mmHg, n=17; p<0.0001;
figure A) with significant lower values in the HDAC4ki ASK mice (p=0.0305). Systolic heart function was only slightly reduced in the ASK group (ASK all: 81.19 ± 3.599%, n=17; Crtl all: 85.95 ± 4.639%, n=17; p=0.0026;
figure B). WT-ASK mice developed mild diastolic dysfunction, displayed by E/E’, while HDAC4ki-ASK mice were completely protected from diastolic dysfunction (E/E’: WT-ASK: -33.35 ± 3.973, n=6; HDAC4ki-ASK: -22.82 ± 1.479, n=11; WT-Crtl: -24.82 ± 2.042, n=8; HDAC4ki-Crtl: -23.03 ± 2.157, n=9; p<0.0001;
figure C). ASK mice displayed a lower heart weight as measured by heart-weight-to-tibia-length ratio (ASK all: 6.583 ± 0.4562mg/mm, n=17; Crtl all: 7.697 ± 0.8121 mg/mm, n=17; p<0.0001;
figure D) with no significant difference between HDAC4ki and WT. Liver and spleen were massively enlarged in the ASK mice, both in absolute organ mass and in relation to body weight and tibia length, also with no difference between HDAC4ki and WT.
 Conclusion
Conclusion This study confirms the protective role of enzymatically inactive HDAC4 in the development of diastolic dysfunction in a L-NAME-independent mouse model with HFpEF. Analyses of atherosclerotic hallmarks are ongoing to reveal the function of HDAC4 in the formation of atherosclerotic plaques. A better understanding of the contribution of the different class IIa HDACs to atherosclerosis can lead to the development of more specific and efficient HDAC inhibitors.



